 三菱ケミカルグループ,慶應義塾大学,日本アイ・ビー・エムは,IBM Quantum Network Hub(慶大量子コンピューティングセンター内)にて「光機能性物質のエネルギーを求めるための量子コンピューターを用いた新たな計算手法」を開発した(ニュースリリース)。
三菱ケミカルグループ,慶應義塾大学,日本アイ・ビー・エムは,IBM Quantum Network Hub(慶大量子コンピューティングセンター内)にて「光機能性物質のエネルギーを求めるための量子コンピューターを用いた新たな計算手法」を開発した(ニュースリリース)。
分子の光機能の理解の深化や合理的設計には,光吸収・発光の波長や強度,発光せずに熱失活してしまう確率などを高精度に求めることが必要。これらの量を求めるには,各現象が最も起こりやすくなる分子構造(FC構造やCI構造)における基底状態と励起状態のエネルギーの定量的な計算が欠かせない。
現在,励起状態の計算にはTDDFT法が最も広く用いられているが,CI構造のように基底状態と励起状態のエネルギー差がない,または小さい場合,エネルギーを正しく求めることができず,代わりに多参照の計算方法を用いる必要があり,膨大な計算コストがかかる。
この問題の解決に量子コンピューターが期待されている。量子コンピューター上で励起状態を計算するためには,基底状態と励起状態を表現できる量子回路と励起状態計算を行なうためのコスト関数を適切に設計する必要がある。
量子コンピューターを用いた計算には,必ず誤差が含まれるため,誤差を最小におさえる量子回路の設計が重要課題だった。また,励起状態計算に適切なコスト関数が,分子構造によって異なるため,すべての分子構造に共通して適用可能な計算方法が求められていた。
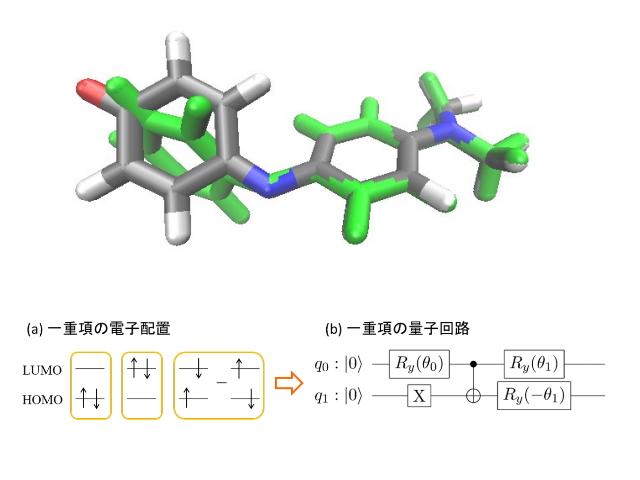
研究では,[A]スピン多重度を保存する量子回路(スピン保存量子回路)の設計指針と[B]コスト関数を用いない励起状態計算法(VQE/AC法)を開発することで,従来よりも小さな計算誤差での励起状態計算に成功した。
実際にスピン保存量子回路とVQE/AC法を組み合わせた多参照計算の一種であるCASSCF法を用いて,フェノールブルー色素の基底状態・励起状態をゲート型量子コンピューターIBM Quantum System One上で計算したところ,FC構造,CI構造いずれの場合も,計算誤差をわずか2kcal/molに抑えてエネルギーを求めることに成功した。
VQE/AC法は励起状態計算のために開発した方法だが,制約条件を変えることで,励起状態に限らず,様々な状態の計算に応用することができるという。また,VQE/AC法を用いることで,任意の分子構造の基底状態・励起状態のエネルギーを同一の計算条件で求められるようになったため,研究グループは,将来的には安定構造や状態間の交差点の構造最適化への応用が期待できるとしている。