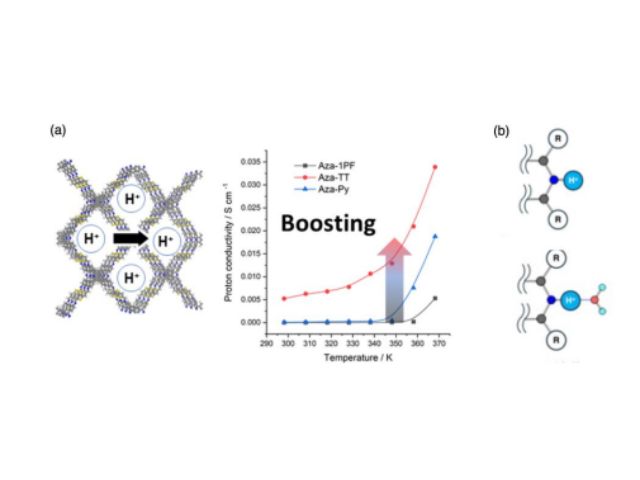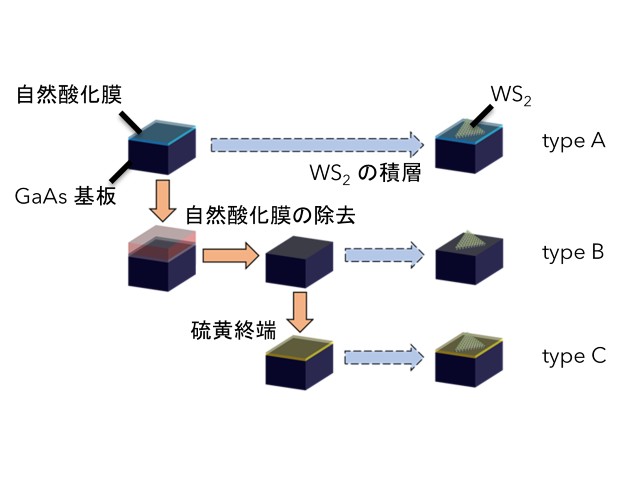
東北大学の研究グループは,二硫化モリブデンに27種類の元素を導入した際の安定な原子構造や電気特性を,密度汎関数理論に基づく精緻な計算機シミュレーションにより明らかにした(ニュースリリース)。
単層が原子3個分の厚さから成る遷移金属ダイカルコゲナイド(TMDC)は,優れた電気特性や光学特性を持つことから,次世代の電子デバイスへの応用が期待されている。特にMoS2は最も精力的に研究がなされているTMDCであり,ウエハースケールでの大面積化を実現していることから,従来のシリコン半導体の限界を超えた,次世代デバイス材料として期待されている。
一方,電界効果トランジスタなどのデバイス製造において,元素を添加するドーピングによって伝導キャリアを導入し,電気特性を制御する必要があることから,MoS2への元素ドーピングに関する研究が盛んに行なわれている。
しかし,実験で用いられる元素は,既にドーピング手法が確立されているReやニオブ(Nb)など数種類に限られている。添加する元素の影響を系統的に調べることができれば,より電気特性の制御に適した元素の発見や,ドーピングによるキャリア導入のメカニズムの正しい理解につながり,結果としてデバイス応用におけるさらなる性能最適化が期待される。
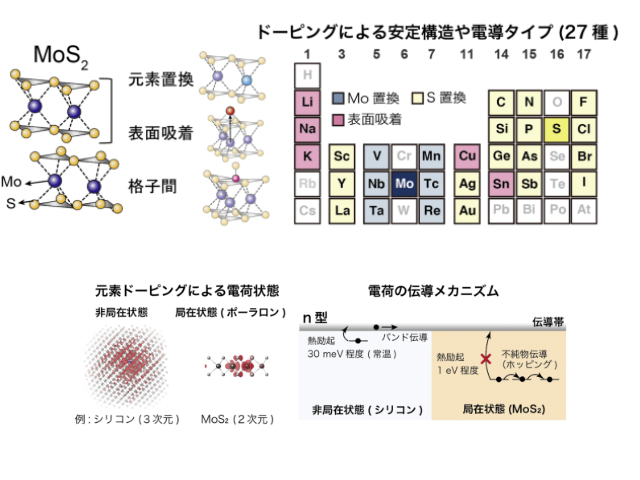
研究グループは,MoS2に導入した27種の元素ドーピングを対象に,密度汎関数理論に基づいた理論計算を行なった。まず,各元素に対し,元素置換,表面吸着,格子間配置など様々な配置を網羅的に計算することで添加元素の安定位置を示し,添加元素ごとの安定な原子構造を明らかにした。
また,シリコンなどの3次元半導体では,伝導キャリアは,空間的に広がった状態で安定となり,室温で容易に励起され伝導に寄与する。しかしMoS2中では,添加元素の種類によらず,空間的に局在するポーラロン状態が安定となり,深い準位を形成することが予測された。
この結果,室温では励起されないことから,実験で観測されている電気伝導は,キャリアが伝導体を移動する通常のバンド伝導とは異なり,添加元素間を飛躍しながら移動する不純物伝導であることが示唆された。
これらの理論予測は,最近実験で観測されているReを添加した際の原子及び電子構造とも良い一致を示しており,予測の確からしさが実験からもサポートされているという。
研究グループは,今後は,他の2次元TMDCへの元素ドーピングに関する理論計算をさらに推進し,より深い知見を得られるように研究を進めていく予定だとしている。