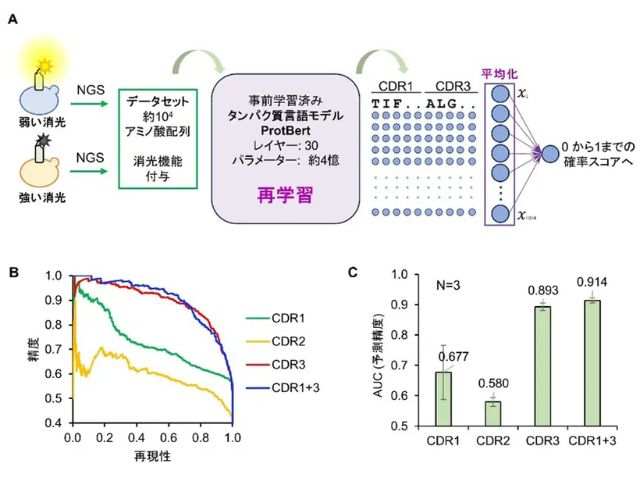
東京工業大学と産業技術総合研究所は,高精度と高速を両立させた最先端の第一原理計算により生成した大規模な理論計算データおよび機械学習を用いて,無機材料表面の基本的な電子構造を網羅的に予測することに成功した(ニュースリリース)。
第一原理計算は,理想的な表面を考慮できることから,イオン化ポテンシャル(IP)と電子親和力(EA)を知るための強力なツールであるが,高精度にIP・EAを算出するには膨大な計算量を必要とする。
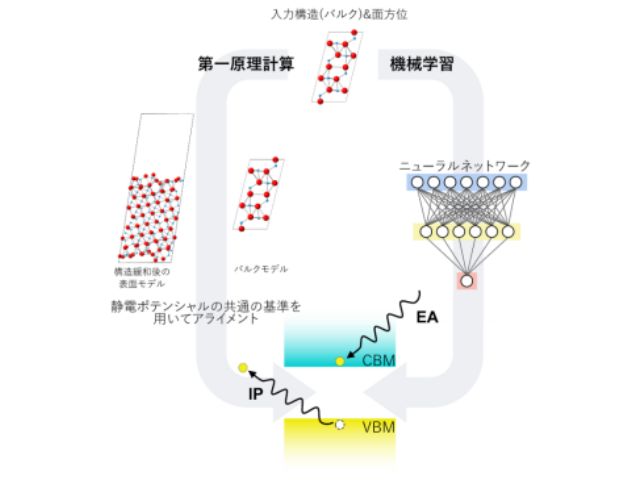
研究グループは,第一原理に基づいた最先端のハイスループット計算手法を用いて,約3,000種類の酸化物表面の原子位置の緩和構造と表面エネルギーおよびIP・EAを計算した。さらに,機械学習による予測モデルを用いて,表面の方位と終端面の位置の情報のみからIP・EAを高精度に予測することを可能にした。
研究グループは,高精度と高速を両立した第一原理計算手法を用いて,まず約2,200種類の二元系酸化物無極性表面のデータベースを構築した。IP・EAの実験値が報告されている酸化物表面を対象に理論計算値と実験値を比較したところ,よく一致していることを確認した。
次に,二元系酸化物表面データベースを用いて,構造緩和前の表面原子配列から構造緩和後のIP・EAを予測するニューラルネットワークを構築した。原子配列の記述子として,ニューラルネットワークに接続することで構成元素の数に対してスケーラブルになるように拡張した固体や分子の原子配列をベクトル化して表現する手法の一つであるSOAPを開発し(L-SOAP),原子配列をベクトル化した。
さらに,三元系酸化物表面への展開を行なった。三元系酸化物は多くの場合,二元系酸化物より複雑な結晶構造を持つことから,その表面について大規模な理論計算データを生成することは難しい。
このため,約700種類の三元系酸化物無極性表面の理論計算データを用意し,二元系酸化物表面について構築したニューラルネットワークをベースとして転移学習を行なった。その結果,予測精度が向上していくことが分かり,今後,三元系酸化物表面の理論計算データが増えることで,さらなる精度改善が期待できるという。
また,通常のSOAPを用いた場合と比べると,L-SOAPを用いて転移学習を行なう方がはるかに予測精度が高く,今回の研究で開発したL-SOAPは転移学習に向いていると言えるという。
研究グループは,この開発された手法は,近年注目されているマテリアルズインフォマティクスに立脚した材料開発を加速することが期待されるとしている。