 大阪市立大学の研究グループは,従来型の最新のコンピュータでも天文学的時間がかかる(指数関数爆発という)分子の全配置間相互作用電子状態計算を量子コンピューター上で現実時間内で計算できる「量子アルゴリズム」を開発し,このアルゴリズムをモデル系に適用しその有効性を実証した(ニュースリリース)。
大阪市立大学の研究グループは,従来型の最新のコンピュータでも天文学的時間がかかる(指数関数爆発という)分子の全配置間相互作用電子状態計算を量子コンピューター上で現実時間内で計算できる「量子アルゴリズム」を開発し,このアルゴリズムをモデル系に適用しその有効性を実証した(ニュースリリース)。
シュレーディンガー方程式は,量子力学の第一原理と言われ,これを厳密に解き,電子のような微視的粒子のどのような情報でも引き出せる関数である波動関数を求めることができれば,分子構造,化学反応経路,分光学的性質,分子物性等を正確に予測することができる。
しかしながら,一電子系など特殊な場合を除き,今日まで分子などの多電子系のシュレーディンガー方程式の厳密解は得ることができず,ざまざま数学的近似法が開発されてきた。
それらの中でも,すべての電子がとりうるすべての配置を考慮する全配置間相互作用法(Full Configuration Interaction Method: FCI法)は,シュレーディンガー方程式の最適解を与える。
ところが,FCI法の計算量は,電子の数及び電子軌道の数に指数関数的に依存するため,二原子分子の窒素N2やシアン(CN)分子のような小さな分子にしか適用できず,これまでFCI法が力を発揮できる環境はなく,その実用性は乏しいと思われてきた。
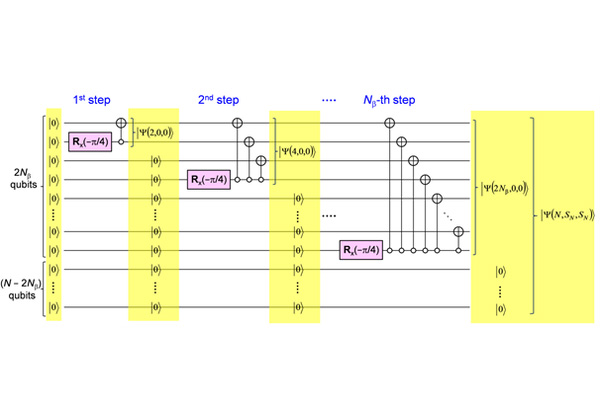
今回,研究グループは、FCI計算を量子コンピューター上で実行できる,簡便な量子アルゴリズムを発見し、FCI計算の指数関数爆発の困難を初めて取り除いた。実際にコンピュータに実装できる量子演算回路を開発し,その有効性を現代のコンピュータを用いて実証した(特許出願中)。
この研究により,量子コンピュータを用いれば,これまで最適解を解くのが最も難しいと思われてきた開殻分子のシュレーディンガー方程式の数値解を効率的に求めることができる可能性が示された。
今までよりも安定かつ小さな単分子磁気メモリーの理論設計,世紀の課題と言われる光合成反応中心に存在するマンガンクラスターの機能解明の量子化学計算など,開殻系が関わる多くの課題はもちろん,創薬分子設計の先行量子化学計算,生体内化学反応解明のためのエネルギー計算時間の大幅な短縮などが期待されるという。
また,今まで実験的に試行錯誤が繰り返されてきた新奇ナノデバイスの設計,新たな熱電変換分子素子の設計などに対しても,理論的なアプローチが可能となる可能性がある。さらに,今回のアルゴリズムは,相対論的効果が表れるさまざまな電磁気学的性質を解明することに適用できるので,相対論的視点に立つ化学などの新領域の開拓に理論面から貢献できるとしている。
関連記事「理研ら,マイクロ波単一光子を高効率に検出」「慶大ら,CNT上に超極細超伝導ナノワイヤを実現」「東大,光子の自在な同期に成功」