 大阪市立大学の研究グループは,量子コンピューターを用いて原子,分子の任意のエネルギー差を直接計算できる量子アルゴリズムを開発した(ニュースリリース)。
大阪市立大学の研究グループは,量子コンピューターを用いて原子,分子の任意のエネルギー差を直接計算できる量子アルゴリズムを開発した(ニュースリリース)。
全配置間相互作用法(full-CI法)と呼ばれる精密な量子化学計算は,従来のコンピュータでは分子サイズに対して指数関数的に計算コストが増大するが,量子コンピュータでは,量子位相推定という量子アルゴリズムを用いることで,分子サイズに対して多項式時間内で計算可能なことが知られている。
しかし,量子コンピュータを用いた量子化学計算では,エネルギー計算値の誤差に反比例して計算コストが増えるため,全エネルギーを小さな桁まで正確に求めることは簡単ではない。
一方で,可視光などの電磁波の吸収波長を計算して分子を同定する,化学反応の進行に必要なエネルギー障壁の高さを求めるなど,量子化学計算で扱う問題のほとんどは分子の全エネルギーそのものではなく,エネルギー差を議論する。
また,大きな分子や周期表で下の方に現れる重原子が入った分子は大きな全エネルギーを持つが,電子励起エネルギーやイオン化エネルギーなど,化学で議論されるエネルギー差の大きさは分子サイズにあまり依存しないという特徴がある。
研究グループは,全エネルギーではなくエネルギー差を量子コンピュータで直接計算することができれば,量子コンピュータを実際の化学研究や物質開発に役立てられると考えた。
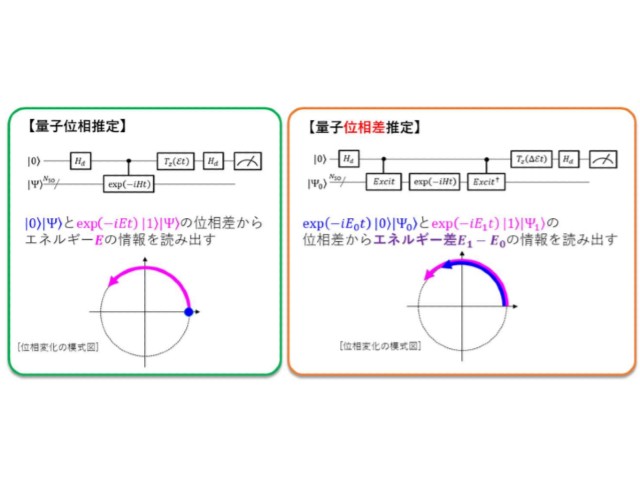
量子コンピュータによるfull-CI計算に用いられる量子位相推定アルゴリズムは,波動関数を時間発展させると,全エネルギーに依存した速さで波動関数の位相が変化する事象を利用する。
これに対し研究グループは,量子位相推定アルゴリズムを応用し,エネルギー差を求めたい2つの電子状態の波動関数の量子重ね合わせ状態を準備し,この量子状態を時間発展させることで,時間発展後の位相差,すなわちエネルギー差を直接計算できる「量子位相差推定」アルゴリズムを開発した。
研究グループはすでに,スピン量子数が異なる電子状態(スピン状態)間のエネルギー差を直接計算する量子アルゴリズムを開発しているが,この量子アルゴリズムでは,①実装に必要な量子ビット数が従来法である量子位相推定よりも増えてしまう,②紫外–可視吸収スペクトルの帰属などで重要となる,スピン量子数が等しい電子状態間の励起エネルギーを求めることができない,といった欠点を抱えていた。
開発したアルゴリズムはこれらの欠点を克服し,汎用性が高いため,量子化学計算だけでなく様々な物理問題・数学問題への応用も期待できるとしている。