 三菱ケミカル,日本アイ・ビー・エム,JSR,慶應義塾大学は,「量子コンピューターを用いた有機EL発光材料の性能予測」の研究プロジェクトで得られた成果に関する論文が,Nature Researchの専門誌「npj Computational Materials」に掲載されたと発表した(ニュースリリース)。
三菱ケミカル,日本アイ・ビー・エム,JSR,慶應義塾大学は,「量子コンピューターを用いた有機EL発光材料の性能予測」の研究プロジェクトで得られた成果に関する論文が,Nature Researchの専門誌「npj Computational Materials」に掲載されたと発表した(ニュースリリース)。
分子の励起状態は,量子コンピューターを用いた励起状態の量子化学計算手法により計算速度が指数関数的に向上し,現在は計算が不可能とされる大きな分子サイズの物質であっても,高精度な計算が可能になると期待されている。
一方でこれら手法のベンチマークテスト計算の多くは,簡単な構造を持つ分子に留まり,より複雑な構造を持つ実用材料への有効性は不明だった。また,量子コンピューター実機の操作エラーの影響を受けるために,実機でこれら手法の計算を実施すると望ましい計算精度は得られないという課題もあった。
研究では量子コンピューターの励起状態計算手法を用いてTADF材料の励起状態エネルギーの計算を行ない,どのように高精度の実機計算を実現できるかに取り組んできた。
TADF材料は最低一重項励起状態(S1)と最低三重項励起状態(T1)の励起状態のエネルギー準位が近いという特性を持ち,その特性を利用して,T1状態にいる励起子が熱運動によりS1状態に遷移すれば,理論上100%の発光効率を有する有機ELの発光材料が得られることになる。そのため,高精度のS1とT1状態の理論計算ができれば,TADF材料の開発を促進すると期待されている。
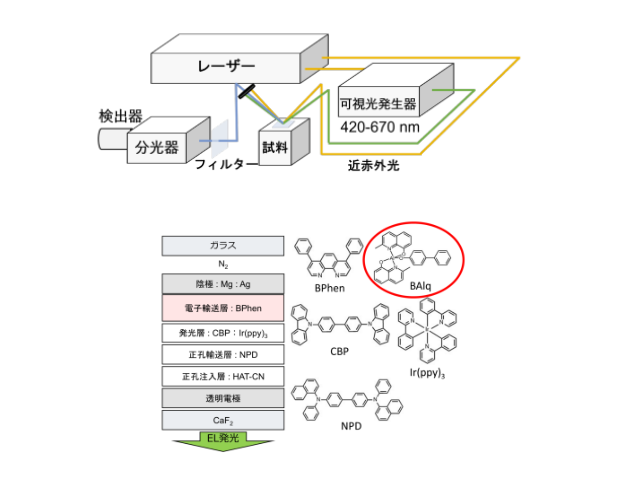
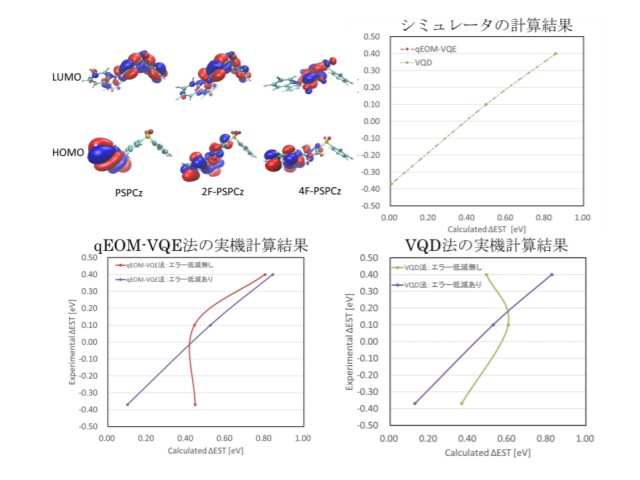
研究では三菱ケミカルの特許で公開されている3種類のTADF材料の分子構造のHOMO,LUMO分子軌道をターゲットにし,これら材料の励起状態計算を行なった。計算は理想的なエラーなしの量子コンピューターシミュレータと現実のIBM量子コンピューター実機で実施した。
シミュレータの計算結果(S1とT1のエネルギーギャップ)は実験値とよい相関(相関係数=0.99)を示すが,実機の計算結果は実機ノイズの影響を受けて実験値と相関性のない結果だった。この実機ノイズによる影響を取り除くために,量子系の一部の記述を特定の実験の測定結果から再構築する実験的な手法である量子トモグラフィーという技術をベースにする実機のエラーを低減する手法を考案した。
この手法を用いたことによって,厳密解と最大88mHaの計算誤差は約4mHaに改善でき,実験値とよい相関の計算値が得られた。研究グループは,今後は開発したエラー低減の手法をさらに発展させ,より大きいサイズの分子軌道の計算を可能にするとしている。