 北陸先端科学技術大学院大学(JAIST)は,物質のミクロな振る舞いをシミュレーションする手法として期待されている「第一原理量子モンテカルロ法」において問題となる計算コストを解消できる手法を見出した(ニュースリリース)。
北陸先端科学技術大学院大学(JAIST)は,物質のミクロな振る舞いをシミュレーションする手法として期待されている「第一原理量子モンテカルロ法」において問題となる計算コストを解消できる手法を見出した(ニュースリリース)。
シミュレーションによる原子運動の予測は,スーパーコンピュータにより大きく進展したが,現在普及しているシミュレーション法では十分な予測精度で原子の運動を計算できない物質群がある。このような問題に対して,大量の乱数を用いた方法を発展させてシミュレーションを行なう「第一原理量子モンテカルロ法」が注目されている。
この手法は,物質の安定性や分子の結合力の大小を比較するため,「エネルギー」の厳密な評価に広く使われてきたが,より進んだ計算である,「原子に働く力」の計算コストが非常に大きいという問題があった。
具体的には,材料に含まれる原子の原子番号が大きくなればなるほど(原子が重くなればなるほど),原子に働く力の計算コストが指数関数的に増えることが問題となっていた。学術界のみならず産業界でも原子レベルでの材料理解は非常に重要視されており,この計算コストの悪化が,第一原理量子モンテカルロ法の普及を妨げていた。
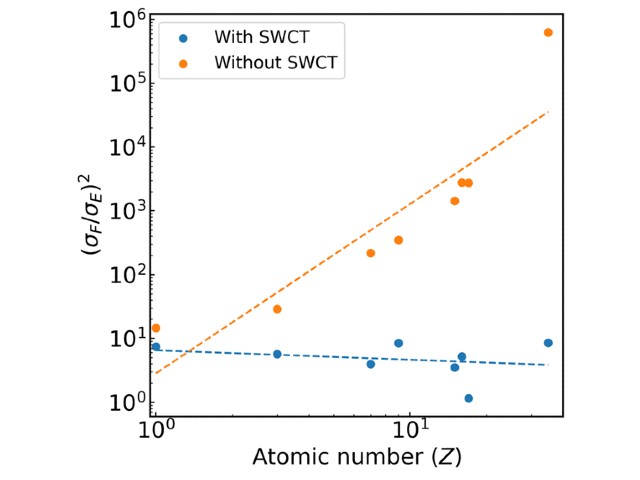
研究グループは,この長らく問題となっていた力の計算コストの悪化を抑えるために,Space-warp coordinate transformation法(SWCT法)を適用することが重要であることを見出した。
SWCT法を適用しない場合,力とエネルギーの計算コストの比が,Z~2.5に従って増加する(Zは原子番号),つまり,「力の計算コストが,重い原子になればなるほどエネルギー計算コストに対して指数関数的に増える」のに対し,SWCT法を適用した場合は,原子番号に関わらず,その比がほぼ定数になることを見出した。
これにより,原子に働く力を以前より高精度かつ短時間で計算できるようになり,材料開発の加速が期待される。なお,この手法は,研究グループらが開発した第一原理量子モンテカルロ法のソフトウェア「TurboRVB」に実装されている。
第一原理量子モンテカルロ法において原子に働く力の計算が確立すると,物質の合成可能性が直接議論できるようになるなど,その応用範囲は劇的に広がる。現在最も普及している原子の電子状態計算は密度汎関数計算だが,応用が進むにつれて,その手法の範囲内では扱えない問題が多数散見されるようになってきているという。
研究グループは開発した手法を用いて,従前法では扱えない物質に対する応用を進めていくとしている。