 大阪大学,三重大学,米ウィスコンシン大学らの研究グループは,磁性分子材料の電子及びスピン状態を高精度に解析・予測するための第一原理計算手法の開発に成功した(ニュースリリース)。
大阪大学,三重大学,米ウィスコンシン大学らの研究グループは,磁性分子材料の電子及びスピン状態を高精度に解析・予測するための第一原理計算手法の開発に成功した(ニュースリリース)。
スピントロニクスを応用した高性能素子の設計・開発には,磁性分子が持つ性質の起源となる電子スピン状態を理解することが不可欠であり,第一原理計算を用いた理論的アプローチが求められる。
一方,その電子スピン状態に関する第一原理計算において,電子軌道の配位子場分裂や電子間にはたらく相関効果の複雑性のために,密度汎関数理論(DFT)の適用に限界があることが知られている。
実際,基底状態の電子スピン状態でさえも,実験と理論計算との両者間で整合性がとれない場合が多く,高い精度での磁性分子材料の理論的設計を行なうためには,これらの障害を克服することが求められていた。
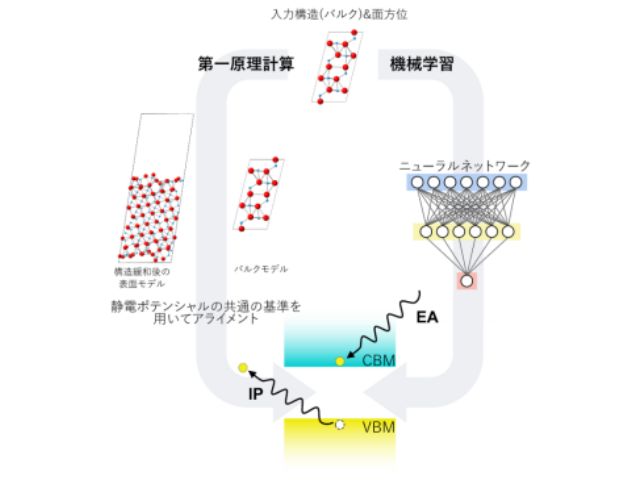
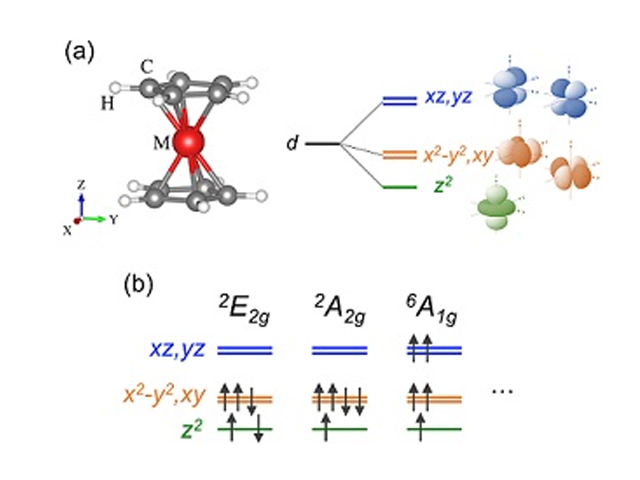
研究グループは,独自に開発してきたFLAPW法を用い,考えられるあらゆる電子スピン状態の全エネルギー計算を行なうことで,高精度に電子スピン状態を解析し,基底状態を探索することができる新たな第一原理計算手法の開発を行なった。
さらに,電子間相関効果の取り扱いを工夫し,相関効果の程度を示す有効オンサイトクーロン相互作用力を定量的に導出し,DFT+U法に基づき多体効果を考慮した。
一般的に,この相互作用力は定性的または経験的に与えられることが多く,それ故,誤った性質の予測に繋がるというリスクがあった。さらに,この手法をメタロセン分子に適用した結果,安定な電子スピン状態やその起源を,世界で初めて,理論的に解明することに成功し,磁性分子材料における種々の物性予測のための新たな計算手法を確立した。
今回の研究は,単一磁性分子の電子スピン状態を高精度に解析し,それに起因する種々の性質や現象を予測することを可能とした理論計算手法を開発したもの。今後,単分子磁石を用いた不揮発性分子メモリや量子コンピュータ等の実用的な磁性分子素子の研究開発への展開が期待されるとしている。